2018.06.08
彰基罕病電子報電子報(第九期)--Mucopolysaccharidoses 黏多醣症
週電子報 第九期 (N20180608) 2018/06/08出刊
1.罕病介紹
◎ ICD-10-CM診斷代碼:
Type1:E76.01 E76.02 E76.03/Type2:E76.1/other:E76.210 E76.211 E76.219 E76.22 E76.29/Unspecified:E76.3 Mucopolysaccharidoses
黏多醣症 ◎
疾病機轉 / 臨床表現
黏多醣症是一組遺傳代謝疾病,其中有缺陷或缺失的?導致大量複雜的糖分子在體內的細胞和組織中以有害的量累積。這種累積會造成永久的,進行性的細胞損傷,影響身體外觀、能力、器官和系統功能,並且在大多數情況下會影響智力發育。身體症狀包括粗糙的面部特徵,嘴唇厚,口腔和舌頭擴大,身材矮小,骨骼尺寸或形狀異常(骨骼不規則),皮膚增厚,肝臟或脾臟等器官增大,疝氣,體毛過度生長、爪狀的手。
流行病學
據估計,在美國出生的每25,000個嬰兒中就有一個會有某種形式的黏多醣症。他們是常染色體隱性遺傳病,意味著只有從雙親遺傳缺陷基因的個體才會受到影響。(MPS II或Hunter綜合徵除外,其中僅母親將缺陷基因遺傳給兒子)。
當一對夫婦中的兩個人都有缺陷基因時,每次懷孕都帶有一次四分之一的機會會受到影響。受影響的孩子的父母和兄弟姐妹可能沒有任何障礙跡象。未受影響的兄弟姐妹和患有黏多醣病的兒童的親屬可攜帶隱性基因,並將其傳遞給自己的孩子。
基因醫學
黏多醣症有許多臨床特徵,但有不同程度的嚴重程度,由於糖胺聚醣的儲存會影響骨骼結構、結締組織和器官,因此這些特徵可能並不明顯。神經系統併發症可能包括神經元損傷、疼痛和運動功能受損。這是由脊髓或周圍神經系統中的神經或神經根的壓縮引起的,神經系統的部分將大腦和脊髓與感覺器官如眼睛和其他器官,肌肉和組織連接起來身體。
受影響的個體可能具有正常的智力或可能嚴重受損,可能經歷發育遲緩,或可能具有嚴重的行為問題。許多人有聽力損失,是傳導性的(耳鼓後面的壓力會導致中耳內層的液體積聚並最終凝結),神經敏感的(內耳中微小的毛細胞受到損傷)或兩者都有。其中腦脊髓液的正常循環隨著時間的流逝而變得阻塞並導致頭部內部壓力增加,這在一些黏多醣貯積病中很常見。通過手術將分流手術插入大腦可排出液體。眼睛的角膜經常從細胞內儲存變得渾濁,漸進性關節僵硬和腕管綜合徵可限制手部活動和功能,反覆的呼吸道感染是常見的,阻塞性氣道疾病和阻塞性睡眠呼吸暫停也是如此,許多受影響的個體也患有心臟病。
遺傳類型
【 以上罕病介紹內容摘錄自National Institutes of Health / 財團法人中華民國
台灣黏多醣症協會】
影音介紹: https://www.youtube.com/watch?v=bINrwv-2oNE
2.彰基諮詢顧問醫師:
兒童遺傳科 --- 趙美琴醫師
3.遺傳診斷現況與發展:
彰化基督教醫院 基因醫學部 研究員/學術副主任 馬國欽 博士
黏多醣症 (Mucopolysaccharidoses)
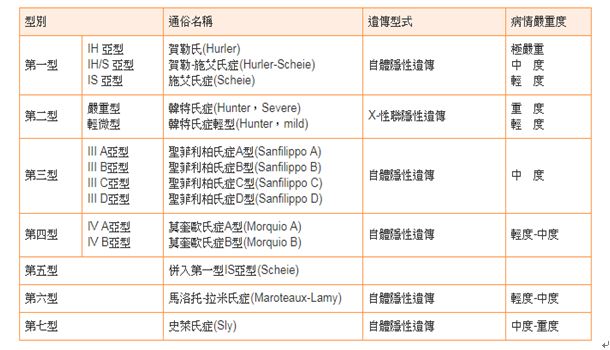
黏多醣症根據缺少的黏多醣水解酵素(Lysosomal hydrolases)種類與患者的臨床症狀,可分為7型 (第一、二、三、四、六、七、九型;其中第五型已併入第一型中的IS亞型,而第八型已經取消),各型之下又可依照病情嚴重程度或是致病基因的不同,區分為不同的亞型 (表一),這其中,除第九型最為少見,目前全球只有一宗病例 (Triggs-Raine et al. 1999),其餘各型平均發生率約在5萬到28萬分之1 (Nelson 1997; Poorthuis et al. 1999),目前全台已知病例超過2百位,但存活個案數不到一半。各型/亞型黏多醣症都是由於單一基因發生缺陷所導致,因此均屬於單基因遺傳疾病,而除了第二型 (MPS II)屬於X-性聯隱性遺傳外,其餘6型的疾病遺傳模式皆為體染色體隱性遺傳。
l X-性聯隱性遺傳: 常由無症狀帶因的母親,將其X染色體上”隱藏不顯露的基因缺陷”傳給男性下一代 (男性X染色體只有一條),致使下一代因無法產生分解黏多醣所需酵素而發病。
l 體染色體隱性遺傳: 常由無症狀分別帶因的父親及母親,同時將各自”隱藏不顯露的基因缺陷”傳給下一代,致使下一代因缺乏合成分解黏多醣所需酵素而發病。
目前黏多醣症多藉由臨床症狀做為診斷參考依據,另外還需進行酵素活性定量與基因檢測作為確認診斷。但由於黏多醣是逐漸累積在細胞內,患者在3歲前身體上可能都不會出現明顯症狀,常導致延宕就診時機。而當發病後,各患者症狀互有差異,且全身各部位也都可能出現症狀,因此就醫時常會先向不同科別(如:兒、骨、內分泌、耳鼻喉等)求診,當最後確診時通常已耗費不少時間。目前透過早期基因篩檢,往往能在患者尚未發病前就驗出黏多醣症,在產前遺傳診斷,則可利用羊膜穿刺術,於懷孕16-20週時,以獲取胎兒細胞進行生化遺傳檢驗或基因檢測,確認胎兒罹病或健康。此外,已知有黏多醣症家族史的家庭,也可以經由人工生殖療程,選擇胚胎植入前遺傳診斷,對胚胎進行遺傳檢測,以篩選健康未罹病胚胎作為植入的對象,避免疾病的再發生與傳遞。
表一、黏多醣症類型、相關基因及遺傳模式。
|
黏多醣症類型
|
疾病通稱/俗稱
|
缺失酵素
|
基因
|
染色體位置
|
累積物質
|
遺傳模式
|
發生率
|
|
MPS第一型 IH亞型
(MPS IH)
|
賀勒氏症 (Hurler syndrome)
|
α-L-iduronidase
|
IDUA
|
4p16.3
|
Heparan sulfate
Dermatan sulfate
|
體染色體隱性
|
1/100,000
|
|
MPS第一型 IH/S亞型
(MPS IH/S)
|
賀勒-施艾氏症 (Hurler-Scheie syndrome)
|
|
MPS第一型 IS亞型
(MPS IS)
|
施艾氏症 (Scheie syndrome)
#舊稱: MPS第五型 (MPS V)
|
|
MPS第二型
(MPS II)
|
韓特氏症 (Hunter syndrome)
|
Iduronate sulfatase
|
IDS
|
Xq28
|
Heparan sulfate
Dermatan sulfate
|
X-性聯隱性
|
1/100,000-150,000 males
|
|
MPS第三型 IIIA亞型
(MPS IIIA)
|
聖菲利柏氏症A型 (Sanfilippo syndrome A)
Sulfamidase缺乏症 (Sulfamidase deficiency)
|
Heparan sulfamidase
|
SGSH
|
17q25.3
|
Heparan sulfate
|
體染色體隱性
|
1/280,000-50,000
|
|
MPS第三型 IIIB亞型
(MPS IIIB)
|
聖菲利柏氏症B型 (Sanfilippo syndrome B)
NAGLU缺乏症 (NAGLU deficiency)
|
N-acetylglucosaminidase
|
NAGLU
|
17q21.2
|
|
MPS第三型 IIIC亞型
(MPS IIIC)
|
聖菲利柏氏症C型 (Sanfilippo syndrome C)
|
Heparan-α-glucosaminide N-acetyltransferase
|
HGSNAT
|
8p11.21
|
|
MPS第三型 IIID亞型
(MPS IIID)
|
聖菲利柏氏症D型 (Sanfilippo syndrome D)
|
N-acetylglucosamine 6-sulfatase
|
GNS
|
12q14.3
|
|
MPS第四型 IVA亞型
(MPS IVA)
|
莫奎歐A型 (Morquio syndrome A)
|
Galactose-6-sulfate sulfatase
|
GALNS
|
16q24.3
|
Keratan sulfate
Chondroitin 6-sulfate
|
|
1/75,000
|
|
MPS第四型 IVB亞型
(MPS IVB)
|
莫奎歐B型 (Morquio syndrome B)
|
β-galactosidase
|
GLB1
|
3p22.3
|
Keratan sulfate
|
|
MPS第五型
(MPS V)
|
#已併入MPS第一型 IS亞型 (MPS IH/S)
|
—
|
—
|
—
|
—
|
—
|
—
|
|
MPS第六型
(MPS VI)
|
馬洛托-拉米氏症 (Maroteaux–Lamy syndrome)
ARSB缺乏症 (ARSB deficiency)
|
N-acetylgalactosamine-4-sulfatase
|
ARSB
|
5q14.1
|
Dermatan sulfate
|
體染色體隱性
|
1/240,000-300,000
|
|
MPS第七型
(MPS VII)
|
史萊氏症 (Sly syndrome)
GUSB缺乏症 (GUSB deficiency)
|
β-glucuronidase
|
GUSB
|
7q11.21
|
Heparan sulfate
Dermatan sulfate
Chondroitin 4,6-sulfate
|
體染色體隱性
|
<1/250,000
|
|
MPS第八型
(MPS VIII)
|
#已取消
|
—
|
—
|
—
|
—
|
—
|
—
|
|
MPS第九型
(MPS IX)
|
Natowicz症 (Natowicz syndrome)
玻尿酸酵素缺乏症 (Hyaluronidase deficiency)
|
Hyaluronidase
|
HYAL1
|
3p21.31
|
Hyaluronic acid
|
體染色體隱性
|
1 case
|
4.營養團隊之建議:
彰化基督教醫院血管醫學防治中心 主任 蔡玲貞
彰化基督教醫院血管醫學防治中心 營養師 麥庭瑜
醫學研究尚未發現有何食物對於各類型黏多醣症孩童有特別助益,所以目前針對臨床病徵給予營養建議。此疾病之患者當有腹瀉情況,應建議避免刺激性食品,包括辣、酸及太甜、油膩、乳製品、含咖啡因食品等;先暫時以低脂肪及低纖維食物為主,注意補充水分及含鈉、鉀電解質液體;細嚼慢嚥並少量多餐,以緩解腹瀉情況。當有便秘情況,可多攝取富含纖維質之食物,包括蔬菜、水果、糙米飯、燕麥等,並且喝足每天每公斤30c.c的水分及適度走路增加腸胃蠕動,可幫助緩解便秘狀況;但隨著病童年齡增長,活動量漸減,肌肉逐漸無力,便秘問題會更加嚴重,如果餐飲或飲品中增加纖維量也不可行時,可請醫師開瀉劑或栓劑。但隨著病情發展,後期患者可能因咀嚼吞嚥動作困難,開始吃東西時會發出聲音或咳嗽,進食時要避免嗆到,最理想的食物為半固體、濃稠流質食物,並且要避免太小(花生、瓜子)、太黏(年糕、湯圓、粽子)等食品。如有熱量攝取不足、過剩或肥胖相關營養問題,皆可進一步諮詢相關專業人士,有助於維持穩定的健康狀態。
5.中醫之建議:
中醫師 邱重閔醫師
黏多醣症病因是體內累積過多的mucopolysaccharides導致一連串的病理變化, 由於小兒即發病, 中醫認為小兒陽氣旺盛, 生長發育會受限主要是這些病理產物的阻礙, 因此治療策略上不能注重補養, 反而要注重排除體內的”瘀毒”, 即過多的mucopolysaccharides, 或用化瘀法, 或用袪痰法, 再酌量使用理氣健脾。有一些醫師經驗分享, 若使用補益藥劑後可能反而產生手足熱、口腔潰瘍、流鼻血等上火現象。畢竟此病為基因遺傳, 而且小兒即發病, 中醫在病程上的伴演角色為改善症狀, 提升生活品質, 也可補充目前西藥不足之處。
檔案下載-彰基罕病電子報電子報(第九期)--Mucopolysaccharidoses 黏多醣症(全文)