2018.08.10
彰基罕病電子報 電子報(第十七期)--結節性硬化症
週電子報 (N20180810) 2018/08/10出刊
1.重要會議
主題:罕見疾病教學及研究會議
日期:107.08.24(五)
時間:14:00-15:30
地點:教研大樓五樓圖書館會議室
內容: 罕病個案研討
罕病專題講座
2.罕病介紹
◎ ICD-10-CM診斷代碼:Q85.1 Tuberous sclerosis 結節性硬化症 ◎
疾病機轉 / 臨床表現
結節性硬化症是一種遺傳性疾病,其特徵在於身體許多部位良性腫瘤的生長。這些腫瘤可能發生在皮膚,大腦,腎臟和其他器官中,在某些情況下會導致嚴重的健康問題。結節性硬化綜合症也會引起發育問題,並且病情的症狀因人而異。
事實上,所有受影響的人都有皮膚異常,包括異常淺色的皮膚斑塊,皮膚凸起和變厚的區域,以及指甲下的生長。面部稱為面部血管纖維瘤的腫瘤在童年時期也很常見。
結節性硬化症常常影響大腦,引起癲癇發作,行為問題,如多動和攻擊性,智力殘疾或學習問題。一些受影響的兒童具有自閉症的特徵,這是一種影響交流和社交互動的發育障礙。良性腦腫瘤也可發生於結節性硬化症患者 ; 這些腫瘤可引起危及生命的併發症。
腎腫瘤在結節性硬化症患者中很常見; 這些增長會導致腎臟嚴重問題功能,在某些情況下可能會危及生命。另外,腫瘤可以在心臟、肺及視網膜中發展。
流行病學
6,000人中有1人會患有結節性硬化症。
基因醫學
TSC1或TSC2基因的突變可導致結節性硬化症。TSC1和TSC2基因提供了用於製備蛋白質錯構瘤蛋白和馬鈴薯球蛋白,在細胞內,這兩種蛋白質可能共同作用,有助於調節細胞生長和大小。這些蛋白質充當腫瘤抑制因子,通常可以阻止細胞生長和分裂太快或以不受控制的方式分裂。
患有結節性硬化症的人在每個細胞中出生時具有TSC1或TSC2基因的一個突變拷貝,這種突變阻止細胞從改變的基因拷貝中產生功能性的hamartin或tuberin。然而,通常從基因的其他正常拷貝產生足夠的蛋白質以有效地調節細胞生長。對於某些類型的腫瘤,在一個人的一生中,必須在某些細胞中發生涉及TSC1或TSC2基因的另一個拷貝的第二個突變。
當TSC1基因的兩個拷貝在特定細胞中突變時,該細胞不能產生任何功能性的hamartin; 具有兩個改變的TSC2基因拷貝的細胞不能產生任何功能性的tuberin。這些蛋白質的缺失使細胞以不受控制的方式生長和分裂,形成腫瘤。在患有結節性硬化症的人中,第二個TSC1或TSC2突變通常發生在受影響的人的一生中的多個細胞中。不同類型細胞中hamartin或tuberin的喪失導致許多不同器官和組織中腫瘤的生長。
遺傳類型
結節性硬化症具有常染色體顯性遺傳模式,這意味著每個細胞中改變基因的一個拷貝足以增加發展腫瘤和其他發育問題的風險。在大約三分之一的病例中,受影響的人從患有該病症的父母那裡繼承了改變的TSC1或TSC2基因,其餘三分之二的結節性硬化症患者出生時都有新的突變在TSC1或TSC2基因中。這些被描述為零星的病例發生在家庭中沒有結節性硬化症病史的人群中。TSC1突變似乎在結節性硬化症體的家族性病例中更常見,而TSC2基因突變在散發病例中更常發生。
【 以上罕病介紹內容摘錄自National Institutes of Health 】
影音介紹:https://www.youtube.com/watch?v=RwxgvDVLqNQ
3.彰基諮詢顧問醫師:
神經醫學部 – 陳大成、吳鴻明
兒童神經科 – 張明裕
皮膚科 – 楊國材
4.遺傳診斷現況與發展:
彰化基督教醫院 基因醫學部 研究員/學術副主任 馬國欽 博士
結節性硬化症(Tuberous sclerosis、Tuberous sclerosis complex (TSC),或稱Epiloia)是一種體染色體顯性遺傳疾病,患者因神經組織細胞和髓鞘形成不良,而在身體不同部位(如: 腦部、肺臟、心臟、腎臟、皮膚、眼睛等)產生結節硬化/結節瘤,常見皮膚症狀,如: 臉部皮膚出現血管纖維瘤或額頭斑塊、身體出現脫色斑或是較為粗糙的鯊魚皮斑,嚴重者可能伴隨癲癇、智能低下、發育遲緩與行為異常等問題,但約有3成的患者擁有正常智力。依據統計,約有1/3患者為家族性遺傳, 2/3個案則為偶發,而TSC在新生兒發生比率約為1/6,000 - 1/10,000 (Randle, 2017)。
TSC依照致病基因的差異可以區分為第一型(TSC1)與第二型(TSC2),其中,TSC1是因為TSC1基因(位於染色體9q34.13)發生缺陷,影響Hamartin蛋白合成所導致;而TSC2則與TSC2基因(位於染色體16p13.3)突變,影響Tuberin蛋白合成有關(表一)。TSC1與TSC2蛋白參與哺乳動物雷帕黴素標靶蛋白訊息傳遞路徑(mTOR signaling pathway)的調控(圖一),正常情況下,TSC1與TSC2蛋白(與另一蛋白TBC1D7)會形成複合體抑制mTOR活性,但當TSC1或TSC2基因發生突變,會影響TSC1與TSC2蛋白調控mTOR蛋白的活性及其後訊號的傳遞,使得蛋白質合成加速,引起細胞不正常連續增生,甚至癌化(Oncogenic transformation)。目前,未有TSC患者在TSC1與TSC2基因上同時發現致病突變,而mTOR signaling pathway中,其他基因(如: TBC1D7、Rheb)上的突變,則都未證實與TSC的發生有關。
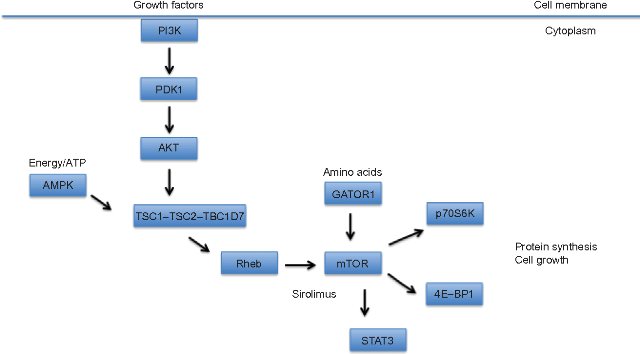
圖一、結節性硬化症與哺乳動物雷帕黴素標靶蛋白訊息傳遞路徑(mTOR
signaling pathway)的調控異常有關(圖片來源:Caban, et al., 2016)。TSC1與TSC2基因上已知具有各式各樣的突變型式,這些突變皆預期會造成轉譯出的蛋白失去功能(Loss of function),或是無法結合形成蛋白質複合體。其中,TSC1較常見的突變型式為小片段缺失/插入(Small deletions/insertions),生產出被截短的蛋白質(Truncated protein),而TSC2的突變型式則涵蓋大片段缺失、小片段缺失/插入、無義突變(Nonsense mutation)與錯義突變(Missense mutation)。TSC1與TSC2突變可以在公開資料庫中(www.lovd.nl/TSC1及www.lovd.nl/TSC2)進行搜尋,截至2018年8月8日,分別有930個TSC1突變與2,689個TSC2突變已經被發表。在整體TSC患者中,TSC2突變較為常見,約佔69%的患者數,而TSC1突變,則約在26%患者中可以發現,此外,有大概5%的患者,其發病原因目前仍不清楚(表一);若單就家族性遺傳的TSC患者而言,TSC1與TSC2突變出現的機率則相對接近(Caban, et al., 2016)。
遺傳檢測對於TSC的確認診斷以及患者的臨床諮詢與處置相當重要,檢測方法多先針對TSC1與TSC2進行全基因定序分析,之後再搭配基因片段缺失/插入分析,如: qPCR、MLPA、aCGH等方法;此外,分子遺傳檢測也可利用多基因檢測套組(Multigene panel)針對複數個基因(如: TSC1與TSC2)進行篩檢。對於家族性個案,則可在產前,甚至胚胎植入前,即針對家族性中已知的基因突變位點進行遺傳檢測或篩檢。
表一、結節性硬化症的分型、相關基因、遺傳模式及遺傳檢測方法。
|
疾病分型
|
基因
|
染色體位置
|
遺傳模式
|
患者佔比*
|
遺傳檢測方法
|
|
結節性硬化症第一型 (TSC1)
|
TSC1
|
9q34.13
|
體染色體顯性
|
~26%
|
1.基因定序分析
2.基因片段缺失/插入分析
3.多基因檢測套組(Multigene panel)分析
|
|
結節性硬化症第二型 (TSC2)
|
TSC2
|
16p13.3
|
體染色體顯性
|
~69%
|
*~5%患者發病原因不明。
5.物理治療之建議:
彰基物理治療師 賴佐君
彰基物理治療師 高倚恩
結節硬化症的主要臨床症狀表現為:癲癇、智能障礙、發育遲緩、精神以及行為問題。物理治療訓練的內容主要包含了知覺動作的訓練、誘發主動動作與強化功能性活動之能力,依據發展順序誘發動作,強化身體控制能力,針對頭部控制及身體抗地心之肌肉的控制、坐姿或站姿的平衡訓練以及行走之功能訓練。而智能障礙除了會有動作發展遲緩、平衡與協調功能較差、體適能與心肺功能較差、步態偏差等問題,也會有注意力缺陷或概念化能力缺陷的問題,因此訓練過程中盡可能簡化任務難度,任務也要具體明確,並透過大量的重複的練習加強孩子類化的能力以達到動作表現的進步。在運動的過程中若癲癇發作,便要立即停止活動且讓個案側躺在安全的地方,鬆開較緊衣物,並視嚴重程度選擇是否要就醫。
檔案下載--彰基罕病電子報 電子報(第十七期)--結節性硬化症(全文)