2018.09.07
彰基罕病電子報電子報(第二十一期)--SCA 脊髓小腦退化性動作協調障礙
週電子報 (N20180907) 2018/09/07出刊
1. 重要會議
時間:9月21日(五) PM2:00 -3:30
地點:教研大樓五樓圖書館 會議室
內容:
罕見研究個案討論
罕病專題講座 -- 先天性痛不敏感症合併無汗症 及 脊髓小腦退化性動作協調障礙2. 罕病介紹
◎ ICD-10-CM診斷代碼:G11.1 脊髓小腦退化性動作協調障礙 Spinocerebellar ataxia ◎
疾病機轉 / 臨床表現
脊髓小腦 共濟失調(SCA)是指一組遺傳性共濟失調,其特徵在於與運動控制相關的大腦部分的退行性變化,有時在脊髓中。有許多不同類型的SCA,它們按照分類突變基因負責特定類型的SCA。使用“SCA”後跟一個數字來描述類型,根據它們的識別順序:SCA1到SCA40。症狀可能因類型而異,可能包括步態不穩,手眼協調能力差和構音障礙。 SCA是一種常染色體顯性遺傳,“脊髓小腦”可能與其他疾病有關,例如常染色體隱性遺傳脊髓小腦性共濟失調(SCAR)。
有許多不同類型的脊髓小腦 共濟失調(SCA)並且每個人都可能有獨特的症狀,受影響的人可能會遇到以下情況:
1.協調和平衡問題(共濟失調)
2.不協調的步伐
3.手眼協調能力差
4.言語異常(構音障礙)
5.不自主的眼球運動
6.視力問題
7.學習和記憶能力受損123 123
流行病學
某些類型的SCA遺傳 在一個 常染色體顯性遺傳 方式是由 三核?酸重複 擴展。三核?酸重複序列是脫氧核糖核酸這種情況重複了很多次。這些重複存在是正常的,它們通常不會引起任何問題。然而,大於正常數量的重複可以乾擾基因的功能,導致遺傳條件。三核?酸重複是不穩定的,並且當從父母傳給孩子時可以改變長度。重複次數的增加通常會導致更早的發病年齡和更嚴重的疾病。
基因醫學
當患有常染色體顯性遺傳病的人有孩子時,每個孩子有50%(1比2)的風險從受影響的父母那裡繼承基因的突變拷貝。
遺傳類型
根據SCA的類型,從兒童期到成年期的任何時候都可以出症狀。 SCA3,也稱為Machado-Joseph病,是最常見的SCA類型。SCA類型9到36是罕見的。預測對於脊髓小腦的人 共濟失調(SCA)各不相同。疾病進展和嚴重程度通常取決於SCA的類型。
SCA的四種最常見的類型:SCA1,SCA2,SCA3和SCA6。受這些類型的SCA影響的人通常在症狀出現後10至15年需要輪椅。許多人最終需要協助執行日常任務。
【 以上罕病介紹內容摘錄自National Institutes of Health 】
影音介紹:https://www.youtube.com/watch?v=zzJj98IdjOY
3.彰基諮詢顧問醫師:
神經醫學部 – 劉青山、王文甫、賴建旭、巫錫霖、蔡定倫
兒童內科 – 張明裕
4.遺傳診斷現況與發展:
彰化基督教醫院 基因醫學部 研究員/學術副主任 馬國欽 博士
脊髓小腦退化性動作協調障礙(Spinocerebellar ataxia,SCA),或稱小腦萎縮症是一群在臨床與遺傳上同具高度歧異性的神經退化性疾病,與小腦退化及周邊神經病變有關,主要特徵為漸進式的運動失調,例如: 步伐不穩、肢體顫抖、手眼不協調等,因此又被稱作「企鵝家族」。SCA平均發生率約萬分0.5至1,但實際盛行率則依地理區及族群的不同而有明顯差異。
SCA的診斷一般靠臨床症狀、家族史,及神經系統檢查,而神經學影像分析(如: 核磁共振成像MRI)與分子遺傳檢測則可進一步用以輔助確診與疾病分型,不同的SCA分型臨床症狀不盡相同,而發病的時間點也有所差異,有些早在兒童期即有明顯症狀,而有些則遲至成年晚期。目前已經知道有超過40種以上的SCA分型,相關基因則超過30個(表一),但仍有部分分型其致病基因並未被證實(如: SCA9),且有些被重複分型(如: SCA15與SCA16)。目前所有SCA分型均為體染色體顯性遺傳,過去所稱的SCA24雖為體染色體隱性遺傳,但現已被重新歸類於體染色體隱性遺傳脊髓小腦退化性動作協調障礙第4型(Autosomal recessive spinocerebellar ataxia 4,SCAR4)(表一)。
依照遺傳變異型式的不同,SCA可大致分為三大類: (1) DNA重複序列的大量擴增(圖一),(2)基因的點突變(Point mutations),(3)長片段DNA的擴增或缺失(表一);而在第(1)大類中,又可依照致病的可能機轉不同再細分為兩類: (1-1)蛋白質轉譯區內(CAG)n重複序列的大量擴增,造成轉譯出的蛋白具有過多麩胺醯酸胺基酸(Polyglutamine,polyQ),影響正常功能,而不正常蛋白的堆積亦可能傷害細胞/組織;(1-2)非蛋白質轉譯區內重複序列的大量擴增,這些擴增的重複序列可導致不正常RNA表現,並與RNA結合蛋白(RNA binding proteins,RBP)結合,造成RBP蛋白質失能及/或RNA積聚變成毒性分子(Paulson, et al., 2017; Ashizawa, et al., 2018)。
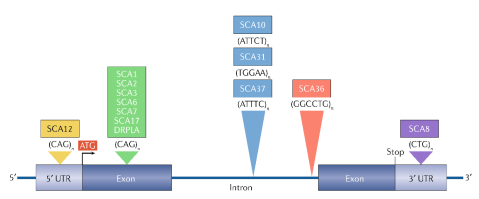
圖一、DNA重複序列的大量擴增是導致脊髓小腦退化性動作協調障礙(Spinocerebellar ataxia,SCA)的主要原因之一。這些重複序列有部分為(CAG)n,造成轉譯出的蛋白具有多麩胺醯酸胺基酸(Polyglutamine,polyQ),而有部分則位於蛋白質非轉譯區(圖片來源:Ashizawa, et al., 2018)。
在SCA各分型中,以SCA1、SCA2、SCA3和SCA6較為常見,其中又以SCA3的發生比率最高,在台灣約佔所有SCA患者的45%,這幾個分型的遺傳變異型式都為DNA重複序列的大量擴增,因此,臨床上對於SCA的初步分子診斷,多先針對這些常見分型,以PCR分子技術進行重複序列擴增次數的分析,若無發現,則再利用次世代定序進行外顯子體序列分析(Exome sequencing),可細部檢測位於外顯子(Exon)及相鄰內含子(Intron)中的遺傳變異,若仍無法確認遺傳變異,則建議再進一步進行全基因體定序分析(Whole genome sequencing),做全面性的遺傳檢測。分子檢測雖可用於SCA的遺傳診斷,但對於部分晚發型的SCA家庭而言,是否要對家族成員都進行檢測,需考量倫理與心理層面的議題,因為受檢者可能在尚無任何症狀時即被告知為罹病高風險,因此,臨床上建議無症狀人員接受SCA篩檢務須先接受遺傳諮詢,以了解檢驗結果所可能造成的衝擊及對後續生活的影響。
表一、脊髓小腦退化性動作協調障礙(Spinocerebellar ataxia,SCA)分型、相
關基因、遺傳變異型式及遺傳模式。
|
疾病分型
|
基因
|
染色體位置
|
遺傳變異型式
|
遺傳模式
|
|
SCA1
|
ATXN1
|
6p22.3
|
CAG repeat expansion
|
體染色體顯性
|
|
SCA2
|
ATXN2
|
12q24.12
|
CAG repeat expansion
|
體染色體顯性
|
|
SCA3
|
ATXN3
|
14q32.12
|
CAG repeat expansion
|
體染色體顯性
|
|
SCA4
|
—
|
16q22.1
|
—
|
體染色體顯性
|
|
SCA5
|
SPTBN2
|
11q13.2
|
Point mutation
|
體染色體顯性
|
|
SCA6
|
CACNA1A
|
19p13.13
|
CAG repeat expansion
|
體染色體顯性
|
|
SCA7
|
ATXN7
|
3p14.1
|
CAG repeat expansion
|
體染色體顯性
|
|
SCA8
|
ATXN8
|
13q21
|
CTG/CAG repeat expansion
|
體染色體顯性
|
|
SCA9
|
—
|
—
|
—
|
體染色體顯性
|
|
SCA10
|
ATXN10
|
22q13.31
|
ATTCT repeat expansion
|
體染色體顯性
|
|
SCA11
|
TTBK2
|
15q15.2
|
Point mutation
|
體染色體顯性
|
|
SCA12
|
PPP2R2B
|
5q32
|
CAG repeat expansion
|
體染色體顯性
|
|
SCA13
|
KCNC3
|
19q13.33
|
Point mutation
|
體染色體顯性
|
|
SCA14
|
PRKCG
|
19q13.42
|
Point mutation
|
體染色體顯性
|
|
SCA15/SCA16
|
ITPR1
|
3p26.1
|
Deletion
|
體染色體顯性
|
|
SCA17
|
TBP
|
6q27
|
CAG repeat expansion
|
體染色體顯性
|
|
SCA18
|
IFRD1
|
7q31.1
|
Point mutation
|
體染色體顯性
|
|
SCA19/SCA22
|
KCND3
|
1p13.2
|
Point mutation, in- frame 3 bp deletion
|
體染色體顯性
|
|
SCA20
|
—
|
11q12
|
260 kb duplication
|
體染色體顯性
|
|
SCA21
|
TMEM240
|
1p36.33
|
Point mutation
|
體染色體顯性
|
|
SCA23
|
PDYN
|
20p13
|
Point mutation
|
體染色體顯性
|
|
SCA24
(現歸類於SCAR4)
|
—
|
1p36
|
|
體染色體隱性
|
|
SCA25
|
—
|
2p21p13
|
—
|
體染色體顯性
|
|
SCA26
|
EEF2
|
19p13.3
|
Point mutation
|
體染色體顯性
|
|
SCA27
|
FGF14
|
13q33.1
|
Point mutation
|
體染色體顯性
|
|
SCA28
|
AFG3L2
|
18p11.21
|
Point mutation
|
體染色體顯性
|
|
SCA29
|
ITPR1
|
3p26.1
|
Point mutation
|
體染色體顯性
|
|
SCA30
|
Candidate gene: ODZ3
|
4q34.3q35.1
|
—
|
體染色體顯性
|
|
SCA31
|
BEAN1
|
16q21
|
TGGAA repeat expansion
|
體染色體顯性
|
|
SCA32
|
—
|
7q32q33
|
—
|
體染色體顯性
|
|
SCA33
|
—
|
—
|
—
|
體染色體顯性
|
|
SCA34
|
ELOVL4
|
6q14.1
|
Point mutation
|
體染色體顯性
|
|
SCA35
|
TGM6
|
20p13
|
Point mutation
|
體染色體顯性
|
|
SCA36
|
NOP56
|
20p13
|
GGCCTG repeat expansion
|
體染色體顯性
|
|
SCA37
|
DAB1
|
1p32.2
|
ATTTC repeat insertion
|
體染色體顯性
|
|
SCA38
|
ELOVL5
|
6p12.1
|
Point mutation
|
體染色體顯性
|
|
SCA39
|
—
|
11q21q22.3
|
7.5Mb duplication
|
體染色體顯性
|
|
SCA40
|
CCDC88C
|
14q32.11q32.12
|
Point mutation
|
體染色體顯性
|
|
SCA41
|
TRPC3
|
4q27
|
Point mutation
|
體染色體顯性
|
|
SCA42
|
CACNA1G
|
17q21.33
|
Point mutation
|
體染色體顯性
|
|
SCA43
|
MME
|
3q25.2
|
Point mutation
|
體染色體顯性
|
|
SCA44
|
GRM1
|
6q24.3
|
Point mutation, +1 bp
frameshift
|
體染色體顯性
|
|
SCA45
|
FAT2
|
5q33.1
|
Point mutation
|
體染色體顯性
|
|
SCA46
|
PLD3
|
19q13.2
|
Point mutation
|
體染色體顯性
|
5.復健治療之建議:
彰基物理治療師 賴佐君
彰基物理治療師 高倚恩
脊髓小腦萎縮症( Spinocerebellar Ataxia, SCA)是一種家族性顯性遺傳的神經系統疾病,動作特徵包含了協同失調、辨距不良、顫抖、輪替運動困難、姿勢性搖晃、平衡不佳、失調性步態。而物理治療的會以「法蘭克氏運動(Frenkel exercise)」來介入協同失調的部分,會先在躺姿下進行肢體的活動,慢慢漸進到坐姿以及站姿;並會在站姿或坐姿下以改變身體的支持底面積以及重心轉移的方式來做訓練;而這類型的病人由於失調性步態會增加跌倒的風險,因此物理治療也會選擇適當的輔具,例如:助行器、四角柺、單柺來輔助,並確實教導病友轉位至輪椅的技巧,以增加日常生活的獨立性。由於脊髓小腦萎縮症是一個不可逆的疾病,目前的醫療介入只能舒緩症狀及減緩惡化進行。小腦細胞老化速度快,須更重視身體的保養,注意飲食起居並配合物理治療的特殊復健訓練,持之以恆的練習,能夠延緩老化達到病情惡化的減緩。

ü 法蘭克氏運動-單腳屈曲伸直

ü 法蘭克氏運動-雙腳運動:一腳曲伸直一腳外展內收
6.營養團隊之建議:
彰化基督教醫院血管醫學防治中心 蔡玲貞 主任
彰化基督教醫院血管醫學防治中心 麥庭瑜 營養師
小腦是中樞神經系統重要的構造之一,控制身體的平衡進而調整姿勢與步伐、動作協調性與肌肉張力的調節等,當小腦產生病變時,將使運動與平衡失調,除了走路不穩,還包括肌肉張力降低、眼球轉動異常、講話模糊不清與吞嚥困難等病徵產生。
吞嚥困難之飲食原則
1. 建議調整飲食質地:因患者吞嚥狀況可將飲食處理為切碎飲食、半流飲食、全流飲食、管灌飲食
2. 增加食物濃稠度:避免稀薄的液體,例如飲料、清湯、茶等;及避免滑溜食物,例如果凍、
仙草、愛玉、粉圓等;且進食使用小湯匙,以避免嗆咳
3. 避免會嗆又不易吞嚥之食品:例如鬆散的酥餅、餅乾、蛋捲等;避免黏稠食品,例如麻糬、
年糕、湯圓、糯米製品等;及避免不易咀嚼完全的食物,例如帶皮、有殼的;
4. 少量多餐。
最後要提醒患者每日均衡六大類飲食,並且適量進食,不受疾病影響,維持健康的飲食狀態。
7.中醫之建議:
彰基中醫部 邱重閔 醫師
小腦萎縮症中醫古名比較相似者為"骨搖", 合併"痿證"或"痺症"等等, 古人雖不知小腦大小, 但以症狀作為疾病分類, 同時也是中醫治療策略的分類。同樣小腦萎縮症, 可能CAG重複序列較少、症狀較輕者, 以肌肉無力作為主要表現; 若CAG repeat重複序列多、症狀較嚴重者, 即以嚴重走路不穩、肌肉僵硬作為表現。痿證(下肢肌肉無力)可能是較輕型的小腦萎縮症, 痺證(肌肉緊繃)可能是較嚴重型的。
小腦神經細胞無法再生, 因此中醫治療策略最好是擺在排除障礙, 儘可能減輕來自身體各方面的氧化壓力, 讓全身的能量能保存為小腦清運垃圾, 才能為小腦保存剩餘的生存空間。治療經驗上, 初期宜排除加重病患退化的因子, 比如睡眠不佳會加重走路不穩、輕飄飄的不踏實感, 即須運用中醫辨證來處方與針灸治療失眠; 若腰背肌群緊繃會加重走路困難, 即須放鬆緊繃肌群。中後期要強化或重建小腦週邊循環, 比如頸椎、上段胸椎、或頭針的針灸刺激, 或用藥上加強補腎填精、滋陰補氣, 以鞏固患者的穩定度。外國文獻亦統計過, 疲勞是這個疾病的一大特色。
外國研究指出高強度的協調性運動對病人的不穩的症狀會改善, 經驗上也聽許多患者回饋, 持續運動比較不會退步。彰基太極拳運動長期以來一直提供此項服務, 並獲病友好評。
不少患者因為對疾病心灰意冷, 不願意持續治療, 因此門診中想儘辦法鼓勵患者持續治療是另一重點, 看過不少失去治療信心的患者退步較為明顯, 而持續運動及治療的維持得較穩定。
檔案下載-彰基罕病電子報電子報(第二十一期)--SCA 脊髓小腦退化性動作協調障礙(全文)