2018.07.20
彰基罕病電子報電子報(第十四期)--ALS 肌萎縮性側索硬化症
週電子報 (N20180720) 2018/07/20出刊
1.罕病介紹
◎ ICD-10-CM診斷代碼:G12.21 Amyotrophic lateral sclerosis 肌萎縮性側
索硬化症 ◎
疾病機轉 / 臨床表現
肌萎縮側索硬化症(ALS)是一種影響運動神經元的進行性疾病,這是控制肌肉運動的專門神經細胞,這些神經細胞存在於脊髓和大腦中。在肌萎縮側索硬化症中,運動神經元隨著時間的推移而萎縮,導致肌肉無力、肌肉量減少,無法控制運動。
有許多不同類型的肌萎縮側索硬化症; 這些類型的特徵在於它們的症狀以及它們的遺傳原因或缺乏明確的遺傳關聯。大多數ALS患者的病情形式被描述為散發性,這意味著它發生在家庭中沒有明顯病史的人群中。一小部分ALS患者,估計為5%至10%,有ALS家族史或稱為額顳葉癡呆(FTD)的相關疾病,這是一種影響人格,行為和語言的進行性腦部疾病。少數情況下,患有家族性ALS的人在兒童期或青少年時期會出現症狀,稱為青少年肌萎縮側索硬化症。
肌萎縮側索硬化最早的症狀包括肌肉抽搐、痙攣、僵硬或虛弱,受影響的個體可能會出現構音障礙,以及吞嚥困難。許多患有ALS的人由於吞嚥困難而導致食物攝入減少,並且由於長期疾病導致他們的新陳代謝增加,因此營養不良。隨著疾病的進展,肌肉變得越來越弱,隨著肌肉組織的萎縮,手臂和腿部看起來變得更薄,最終會失去肌肉力量和行走能力。受影響的個人最終變得依賴輪椅,並越來越需要個人護理和其他日常生活活動的幫助。大多數ALS患者在出現ALS症狀後2至10年內死於呼吸衰竭。
大約20%的ALS患者也患有FTD,人格和行為的改變可能使受影響的個人難以適合社會的方式與他人互動,隨著疾病的進展,溝通技巧會惡化;尚不清楚ALS和FTD的發展是如何相關的,發生這兩種病症的個體被診斷為患有ALS-FTD。
通常在家族中運行的罕見形式的ALS被稱為ALS-帕金森病 - 癡呆綜合症(ALS-PDC)。除了稱為帕金森病的運動異常模式和癡呆的進行性喪失之外,這種疾病的特徵在於ALS的症狀。帕金森症的症狀包括運動遲緩、僵硬和震顫。同一家庭的受影響成員可能具有不同的症狀。
流行病學
美國每年約有5,000人被診斷患有肌萎縮側索硬化症。在全球,這種疾病發生在每10萬人中2至5人。只有一小部分病例具有已知的遺傳原因。
在關島的查莫羅人和日本紀伊半島的人中,ALS-PDC的頻率是ALS在其他人群中的100倍。外。
基因醫學
幾種基因的突變可引起家族性ALS,並有助於散發性肌萎縮側索硬化的發展。在美國和歐洲,C9orf72基因的突變佔家族性ALS的30%至40%。在全球,SOD1基因突變導致15%至20%的家族性ALS,並且TARDBP和FUS基因突變各佔約5%的病例。與家族性肌萎縮側索硬化症相關的其他基因各佔一小部分病例。據估計,60%的家族性ALS患者具有已確定的基因突變。其餘個體病情的原因尚不清楚。
C9orf72,SOD1,TARDBP,和FUS基因是關鍵的正常運作馬達的神經元和其他細胞。目前尚不清楚這些基因的突變是如何導致運動神經元死亡的,但人們認為運動神經元由於體積龐大而對功能中斷更為敏感。受ALS影響的大多數運動神經元具有聚集體的累積; 然而,尚不清楚這些聚集體是否參與引起ALS或是垂死細胞的副產物。
在一些其他基因突變引起的家族性ALS病例中,研究已經確定了導致ALS的機制。一些基因突變導致軸突發育的破壞,傳遞神經衝動的運動神經元的擴展。改變的軸突可能損害從神經到肌肉的脈衝傳遞,導致肌肉無力和萎縮。其他突變導致運動神經元中軸突正常功能所需的材料運輸減慢,最終導致運動神經元死亡。額外的基因突變可以防止有毒物質的分解,從而導致它們在神經細胞中積聚。有毒物質的積累會損害運動神經元並最終導致細胞死亡。在某些ALS病例中,基因突變如何導致病情尚不清楚。
散發性肌萎縮側索硬化的原因還未知,但可能涉及遺傳和環境因素。許多基因的變異,包括前面提到的涉及神經衝動傳遞和神經元內物質運輸的基因,增加了發生肌萎縮側索硬化的風險。作為ALS危險因素的基因突變可能會添加,刪除或改變核?酸,導致產生具有改變或降低功能的蛋白質。雖然遺傳變異與散發性肌萎縮側索硬化有關,但並非所有遺傳因素都已被確定,並且尚不清楚大多數遺傳變化如何影響疾病的發展。
遺傳類型
大約90%至95%的ALS病例是散發性的,這意味著它們不是遺傳的。
據估計,5%至10%的ALS是家族性的,並且由幾種基因之一的突變引起。遺傳模式取決於所涉及的基因。大多數病例以常染色體顯性遺傳方式遺傳,這意味著每個細胞中一個改變基因的拷貝足以引起疾病。在大多數情況下,受影響的人有一個父母患有該病症。一些遺傳已知導致ALS的家族性基因突變的人從未發展出這種疾病的特徵(這種情況被稱為減少外顯率)。目前尚不清楚為什麼一些有突變基因的人會患上這種疾病,而另一些有突變基因的人卻沒有。
不太常見的是,ALS以常染色體隱性模式遺傳,這意味著每個細胞中基因的兩個拷貝都有突變。具有常染色體隱性病症的個體的父母每個攜帶一個突變基因的拷貝,但它們通常不顯示該病症的症狀。由於受影響的人的父母不受影響,常染色體隱性遺傳性ALS常被誤認為是散發性ALS,即使它是由家族性基因突變引起的。
ALS以X連鎖顯性模式遺傳,當與該病症相關的基因位於X染色體上時,發生X連鎖狀況,X染色體是兩個性染色體之一。在雌性(具有兩條X染色體)中,每個細胞中基因的兩個拷貝之一中的突變足以引起該疾病。在雄性(只有一條X染色體)中,每個細胞中唯一一個基因拷貝的突變會導致這種疾病。在大多數情況下,與女性相比,男性更容易患病,預期壽命縮短。X連鎖遺傳的一個特徵是父親不能將X連鎖特徵傳遞給他們的兒子。
【 以上罕病介紹內容摘錄自National Institutes of Health 】
影音介紹:https://www.youtube.com/watch?v=arDpeOm2s_4
2.彰基諮詢顧問醫師:
神經醫學部 -- 劉青山、巫錫霖、賴建旭、莊介森
3.遺傳診斷現況與發展:
彰化基督教醫院 基因醫學部 研究員/學術副主任 馬國欽 博士
肌萎縮性側索硬化症(Amyotrophic lateral sclerosis, ALS),俗稱漸凍人,或稱路‧蓋里格氏症(Lou Gehrig’s disease),是一種運動神經系統退化性疾病,起因於身上負責傳遞收縮訊息給肌肉的運動神經元(Motor neuron)逐漸凋亡,致使肌肉漸漸萎縮與無力,常侵犯腦部或脊髓,病程漸進但快速,一般發病後2-5年內患者即失去對肌肉的控制能力而完全癱瘓,終至死亡。
ALS診斷主要仰賴病史、神經學檢查以及其他客觀性檢查,一般常見病理特徵包括同側肢體的下運動神經元徵象(如: 無力、萎縮和肌束抽搐)及上運動神經元徵象(如: 肌腱反射上升、霍夫曼徵象(Hoffmann signs)、巴賓斯基反射(Babinski signs)或肌躍症)。
ALS多為偶發性且病因不明,環境因素、遺傳因素、重金屬中毒、病毒感染,與自體免疫異常等,都被懷疑是可能的致病因子。大約有5-10%的ALS則是屬於家族性遺傳,意即遺傳自雙親,與ALS有關的分子遺傳途徑相當多元(圖一),已知基因則超過30個(Chen, et al. 2013; Nguyen, et al. 2018),遺傳模式涵蓋體染色體顯性、體染色體隱性,及X-性聯顯性,疾病本身則可依照致病基因及遺傳模式進行分型(表一)。ALS在遺傳方式另有一特點,即偏向寡基因遺傳的型式(Oligogenic mode of inheritance),也就是通常要2個或多個基因同時發生變異才會引起疾病。
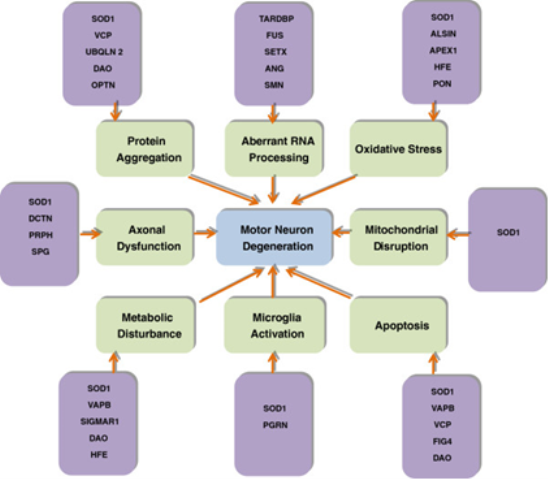
圖一、肌萎縮性側索硬化症可導因於與運動神經元有關的各類分子遺傳途徑相互
影響及鄰近非神經細胞間的交互作用(圖片來源:Chen et al., 2013)。
分子遺傳檢測可用於ALS診斷與分型,依據家族史、遺傳模式及相關基因在患者族群中出現的頻率,可以選定特定基因先進行序列分析,其中,C9orf72是最常被關聯的ALS基因,約有40%家族性遺傳個案及7%偶發性個案都與該基因變異有關;SOD1則是其次與ALS關聯的基因,約12%家族性遺傳個案及1-2%偶發性個案與該基因變異有關;TARDBP與FUS基因變異則都見於約1-5%家族性遺傳個案及<1%偶發性個案。此外,分子遺傳檢測也可直接針對多個基因進行篩檢,但考量不同基因其常見突變類型可能不同,因此,不同ALS患者可能需要不同的多基因檢測套組(Multigene panel)與對應的檢測方法。
此外,ALS屬晚發型疾病,偶發性與家族性遺傳個案發病平均年齡約分別在56歲及46歲,分子檢測雖可提早發現家族中高風險個案,但診斷過程卻可能涉及倫理議題,主要是ALS具有不完全外顯(Incomplete dominance)特性,以及受檢者可能在尚無任何症狀時即被告知可能罹病,影響後續生活,另外,現階段ALS並無有效的治療方法,因此,遺傳檢測用於家族成員ALS篩檢需特別小心,務必接受遺傳諮詢,尤其是在未成年人身上。但已知有ALS家族史之夫妻,則可考慮在計畫懷孕前即進行胚胎植入前遺傳診斷。
表一、肌萎縮性側索硬化症(Amyotrophic lateral sclerosis, ALS)分型、相關
基因及遺傳模式。FTD, frontotemporal dementia。
|
疾病分型
|
基因
|
染色體位置
|
遺傳模式
|
|
ALS1
|
SOD1
|
21q22.1
|
1.體染色體顯性
2.體染色體隱性
|
|
ALS2
|
ALS2
|
2q33.1
|
體染色體隱性
|
|
ALS3
|
未知
|
18q21
|
體染色體顯性
|
|
ALS4
|
SETX
|
9q34.13
|
體染色體顯性
|
|
ALS5
|
SPG11
|
15q21.1
|
體染色體隱性
|
|
ALS6
|
FUS
|
16p11.2
|
1.體染色體顯性
2.體染色體隱性
|
|
ALS7
|
未知
|
20p13
|
體染色體顯性
|
|
ALS8
|
VAPB
|
20q13.3
|
體染色體顯性
|
|
ALS9
|
ANG
|
14q11.2
|
體染色體顯性
|
|
ALS10
|
TARDBP
|
1p36.2
|
體染色體顯性
|
|
ALS11
|
FIG4
|
6q21
|
體染色體顯性
|
|
ALS12
|
OPTN
|
10p13
|
1.體染色體顯性
2.體染色體隱性
|
|
ALS13
|
ATXN2
|
12q24.12
|
體染色體顯性
|
|
ALS14
|
VCP
|
9p13.3
|
體染色體顯性
|
|
ALS15
|
UBQLN2
|
Xp11.21
|
X-性聯顯性
|
|
ALS16
|
SIGMAR1
|
9p13.3
|
體染色體隱性
|
|
ALS17
|
CHMP2B
|
3p11.2
|
體染色體顯性
|
|
ALS18
|
PFN1
|
17p13.2
|
體染色體顯性
|
|
ALS19
|
ERBB4
|
2q34
|
體染色體顯性
|
|
ALS20
|
HNRNPA1
|
12q13.13
|
體染色體顯性
|
|
ALS21
|
MATR3
|
5q31.2
|
體染色體顯性
|
|
ALS22
|
TUBA4A
|
2q35
|
體染色體顯性
|
|
ALS23
|
ANXA11
|
10q22.3
|
體染色體顯性
|
|
ALS24
|
NEK1
|
4q33
|
未知
|
|
ALS25
|
KIF5A
|
12q13.3
|
體染色體顯性
|
|
FTD-ALS1
|
C9orf72
|
9p21.2
|
體染色體顯性
|
|
FTD-ALS2
|
CHCHD10
|
22q11.23
|
體染色體顯性
|
|
FTD-ALS3
|
SQSTM1
|
5q35.3
|
體染色體顯性
|
|
FTD-ALS4
|
TBK1
|
12q14.2
|
體染色體顯性
|
|
IBMPFD2
|
HNRNPA2B1
|
7p15.2
|
體染色體顯性
|
|
未分型1
|
C21orf2
|
21q22.3
|
未知
|
|
未分型2
|
CCNF
|
16p13.3
|
體染色體顯性
|
|
未分型3
|
TIA1
|
2p13.3
|
體染色體顯性
|
4.營養團隊之建議:
彰化基督教醫院血管醫學防治中心 蔡玲貞 主任
彰化基督教醫院血管醫學防治中心 麥庭瑜 營養師
肌 萎 縮 側 索 硬 化 症 ( amyotrophic lateral sclerosis, ALS )是一種退化性神經疾病,隨著疾病發展,語言、吞嚥以及呼吸功能都會受到影響。其中吞嚥困難會使ALS患者的食物攝入量減少,且此疾病長期會使患者新陳代謝增加,最終將導致營養不良。ALS 病人營養處置的目標為維持足夠營養、預防吸入性肺炎,所以應即早給予營養介入,避免患者體重下降。
因ALS患者的能量消耗約增加10-20%,為維持足夠的營養,熱量建議為35 kcal/kg、蛋白質需要量建議為1-1.5 g/kg/day,不建議少於1.0 g/kg/day。ALS患者因飲食攝取不足及曝曬陽光時間少,容易造成骨質疏鬆症,所以建議可另外補充維生素D。另外已有研究指出補足體內維他命E濃度可有效降低疾病之進展和患有ALS的風險。ALS患者主要是因延髓(bulbar)逐漸功能退化而導致吞嚥困難,患者臨床症狀包括進食易嗆咳、咀嚼能力降低、進食速度慢、唾液分泌增加等,所以飲食供應建議包括選擇質地易消化的食物、液體適時添加增稠劑、補充高熱量高蛋白食物、防止刺激性食物等。由於患者吞嚥困難、減少食物攝取量、增加吸入性肺炎發生頻率,必要時需要考慮改變進食途徑,短期營養支持可使用鼻胃管灌食;長期營養支持可使用經皮內視鏡下做胃造口術;為避免患者營養狀況不佳,應盡早評估進食途徑,降低患者呼吸衰竭和死亡率的風險。
5.中醫之建議:
彰基中醫部 邱重閔 醫師
退化性的肌肉無力、活動困難在中醫病名為”痿症”, 內經提到”治痿獨取陽明”意思是治療這類疾病, 要重視脾胃功能、也就是消化系統功能要治療好; 內經還有還有提到”五臟因肺熱葉焦, 發為痿躄”, 後世各朝代醫家陸續提出”五臟皆有痿”, 意思是五臟(肝-自律神經系統、心-心血管系統、脾-消化系統、肺-呼吸系統+免疫系統、腎-生殖系統+免疫系統)都可能造成或加重痿症, 所以診斷上無法一概而論, 治療上也沒有專病特效藥。
總結臨床醫案報導與個人治療經驗, 患者主要症狀為神經退化, 針灸治療必須著重督脈, 也就是脊椎與顱腦部位的刺激, 強化神經細胞活化, 第二重點在排除其他干擾障礙, 比如減少身體氧化壓力, 改善腸胃功能狀態, 改善睡眠, 改善泌尿系統症狀(比如有頻尿.夜尿等), 惟有減少身體其他地方氧化壓力, 才能使身體專心修復神經退化病灶處。
最後必須結合患者心理狀態鼓勵, 良好的配合治療能減緩病程, 但許多患者常消極自我放棄, 此時若醫療團隊配合心靈鼓舞, 相信能對病程有所助益。
檔案下載-彰基罕病電子報電子報(第十四期)--ALS 肌萎縮性側索硬化症